Chaoxun Wang
Отделение эндокринологии, Пудунская больница Шанхая, Пудунский медицинский центр при Фуданьском университете, Китай
Резюме
Сахарный диабет (СД) 2 типа тесно связан с дисфункцией щитовидной железы. В литературе акцентируется внимание на связи между аномалиями гормонов щитовидной железы и СД 2 типа. Наиболее вероятным механизмом, который ведет к развитию СД 2 типа при дисфункции щитовидной железы, является нарушение генетической экспрессии ряда генов наряду с физиологическими отклонениями, провоцирующими нарушения утилизации глюкозы, повышенной выработки глюкозы печенью и усиленное поглощение висцеральной глюкозы. Эти факторы способствуют инсулинорезистентности. Резистентность к инсулину также связана с дисфункцией щитовидной железы. Гипертиреоз ассоциирован с инсулинорезистентностью, о чем говорят случаи нарушения метаболизма глюкозы при СД 2 типа. Огромное количество доклинических, молекулярных и клинических исследований подтвердило неоспоримую роль дисфункции щитовидной железы как сопутствующего расстройства при СД 2 типа.
Вступление
В 1927 году Coller и Huggins исследовали роль гипертиреоза при сахарном диабете (СД) и доказали наличие связи между гипертиреозом и отягощенным течением диабета. Так, эти исследователи продемонстрировали, что частичное хирургическое удаление щитовидной железы оказывало благоприятный эффект на восстановление толерантности к глюкозе у пациентов с гипертиреозом, болеющих СД [1].
Существует глубинная основополагающая связь между СД и нарушением функции щитовидной железы [2]. Огромное количество исследований выявили целый ряд сложных связанных между собой биохимических, генетических и гормональных нарушений, отражающих эту патофизиологическую связь [2, 3]. Активируемая 5' аденозинмонофосфатом протеинкиназа (АМФК) представляет собой центральную мишень, на которую направлен процесс регуляции чувствительности к инсулину и обратной связи с тиреоидными гормонами, связанной с аппетитом и расходом энергии [3]. Гиперактивность щитовидной железы (болезнь Грейвса/Базедова болезнь) изучалась на предмет ее связи с СД. Проведенный метаанализ показал, что в 11 % случаев у пациентов с СД отмечалась дисфункция щитовидной железы [4]. Аутоиммунная реакция подозревалась в числе главных причин развития СД, связанного с дисфункцией щитовидной железы [5-7].
Нелеченый предиабет (нарушения метаболизма, предшествующие диабету) как 1, так и 2 типа, может вызывать развитие «состояния пониженного Т3», которое характеризуется пониженным сывороточным уровнем общего и свободного Т3, повышенным уровнем обратного Т3 (обТ3) и почти нормальными сывороточными концентрациями Т4 и тиреотропного гормона (ТТГ) [8]. Взаимосвязь между СД 2 типа и тиреоидной дисфункцией менее изучена и может включать в себя ответы на вопросы, связанные с различными аспектами метаболического синдрома, например, с атеросклерозом, артериальной гипертензией и сопутствующими сердечно-сосудистыми заболеваниями.
Эпидемиология
Дисфункция щитовидной железы (тиреоидная) – распространенная эндокринная патология с неравномерной распространенностью. Исследование, проведенное Wickham, показывает, что распространенность тиреоидной дисфункции у взрослых мужчин в Англии составляет 6,6 % [11]. По результатам Колорадского исследования, у 9,5 % участников был обнаружен повышенный, а у 2,2 % – пониженный уровень ТТГ. Данные Национальной программы проверки здоровья и питания (исследование NHANES III) показывают, что гипертиреоз и гипотиреоз были выявлены у 4,6 % и 1,3 % от общего количества участников соответственно [12]. Распространенность тиреоидной дисфункции растет с возрастом во всем мире, причем эти показатели у женщин были выше, чем у мужчин. Распространенность субклинического гипертиреоза достигает порядка 2 %.
Заболеваемость тиреоидными нарушениями у больных СД составляет 13,4 %, причем у женщин с СД 2 типа отмечалась более высокая распространенность (31,4 %), чем у мужчин (6,9 %) с СД 2 типа [13]. По данным Akbar и соавт. [14], распространенность тиреоидной дисфункции у пациентов с СД 2 типа в Греции составляла 12,3 %, а в Саудовской Аравии – 16 %. Таким образом, достаточно много пациентов с СД 2 типа были более предрасположены к заболеваниям щитовидной железы.
Периферические эффекты тиреоидных гормонов на секрецию и чувствительность к инсулину
Тиреоидные гормоны напрямую контролируют секрецию инсулина. При гипертиреозе – более выраженный ответ бета-клеток на глюкозу и катехоламины по причине увеличения массы бета-клеток. Кроме того, при тиреотоксикозе повышается клиренс инсулина [15, 16].
Тиреотоксикоз
Центральной причиной, ведущей к гиперинсулинемии, развитию нарушения толерантности к глюкозе и инсулинорезистентности периферических тканей, считается повышенная продукция глюкозы в печени [17]. Толерантность к глюкозе при тиреотоксикозе обусловлена усиленной продукцией глюкозы в печени наряду с активированным гликогенолизом [2]. Именно этот феномен лежит в основе отягощенного течения субклинического диабета и усугубления гипергликемии при СД 2 типа. Тиреотоксикоз может привести к развитию кетоацидоза, в том числе и в силу усиленной липолитической активности и повышенного бета-окисления в печени [18, 19]. Этот феномен показан на рисунке 1.
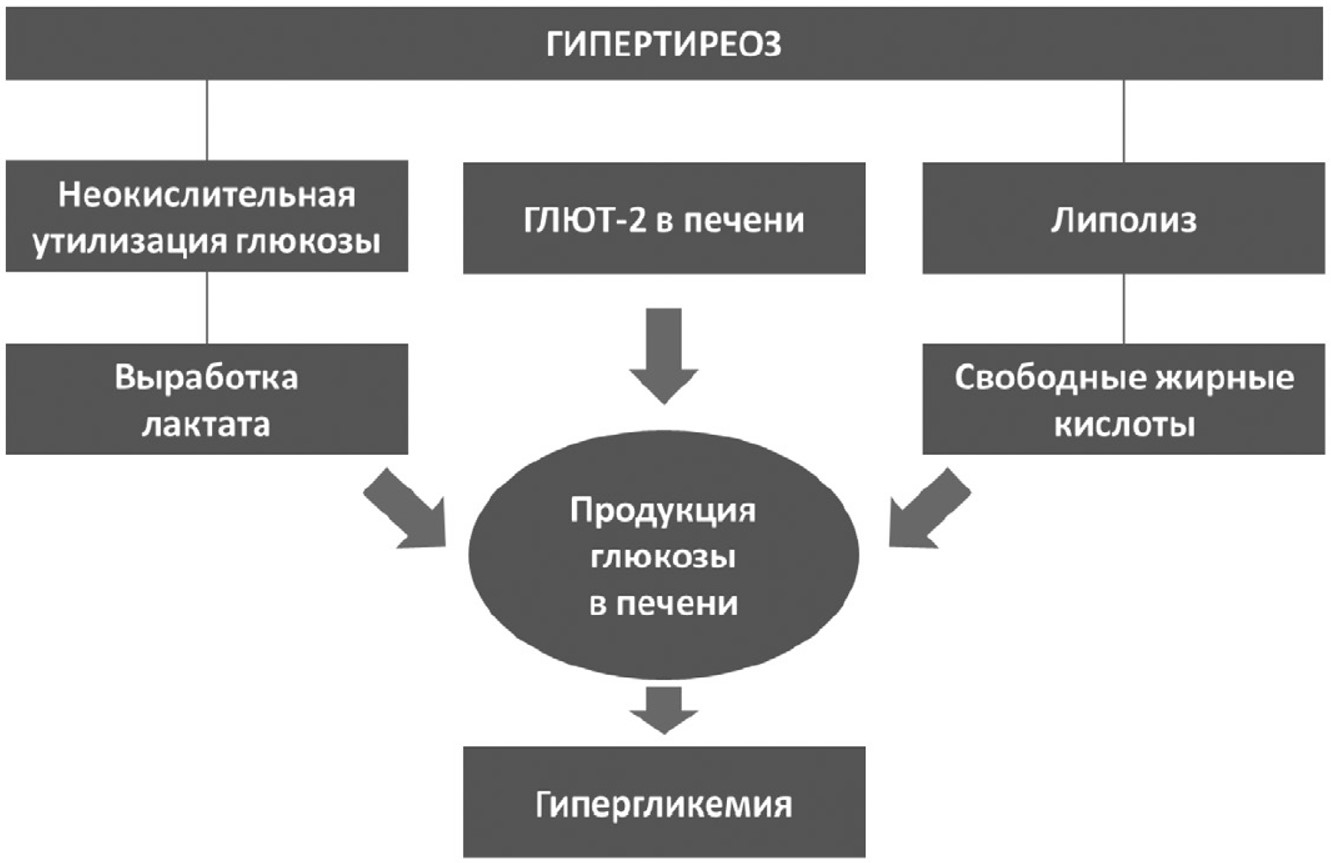
Рис. 1. Взаимосвязь между гипертиреозом и гипергликемией, обусловленная метаболизмом липидов, окислительным стрессом и печеночной дисфункцией
Примечание: ГЛЮТ-2 ‒ глюкозный белок-транспортер 2 типа.
|
Клинический случай 1 Пациент ‒ мужчина 30 лет с компенсированным сахарным диабетом 1 типа, болеет 20 лет, (HbA1с <8 %, норма 5,5-7,7 %), без эпизодов ДКА в анамнезе, в течение месяца чувствует себя плохо, на фоне чего ухудшился гликемический контроль. Органные диабетические осложнения отсутствуют, курит по 25 сигарет в день, но алкоголь не употребляет. Урологические симптомы, грипп, инфекцию дыхательных путей и диарею отрицает. Пациент находился в состоянии обезвоживания и шока, артериальное давление 100/70 мм рт. ст., частота сердечных сокращений 130 уд/мин, но температура тела не повышена. Отмечались тяжелая глюкозурия и кетонурия, уровень глюкозы в капиллярной крови ‒ 28 ммоль/л, HbA1с ‒ 11,8 %, рН артериальной крови ‒ 7,17; бикарбонат крови ‒ 8 ммоль/л. Развернутый общий анализ крови, печеночные и почечные показатели, электролиты сыворотки (калий 5,1 ммоль/л), рентгенография грудной клетки и электрокардиограмма в норме. Бакпосевы крови и мочи стерильны. Диагностирован ДКА, лечение было стандартным: внутривенное введение жидкостей и инсулина, также проводилась эмпирическая антибиотикотерапия. Состояние пациента улучшалось очень медленно, в первые сутки ему потребовались 180 Ед инсулина (обычно 56 Ед/день). Сохранялась тахикардия (120-130 уд/мин), отмечался мелкий тремор, был выявлен зоб 2-4 степени. Подозрение на тиреотоксикоз подтвердилось биохимическими анализами: тиреотропный гормон (ТТГ) <0,06 мЕд/л, общий тироксин ‒ 252 нмоль/л. Антитела к тиреоидной пероксидазе положительные. На 3-й день начата терапия КАРБИМАЗОЛОМ в дозе 40 мг в сутки перорально и пропранололом в дозе 80 мг в день. Пациент быстро пошел на поправку и через 10 дней был выписан домой. От редакции Есть данные, что КАРБИМАЗОЛ имеет наиболее благоприятный профиль безопасности Antithyroid drug regimen for treating Graves’ hyperthyroidism (Review) Abraham P, Avenell A, McGeoch SC, Clark LF, Bevan JS The Cochrane Library 2010, Issue 1 |
Взаимосвязь между патологическими проявлениями гипертиреоза и сахарного диабета 2 типа: роль инсулинорезистентности и других факторов
К патологическим признакам СД 2 типа относятся повышенная абсорбция глюкозы в желудочно-кишечном тракте, пониженная секреция инсулина и изменения в массе бета-клеток [22]. Помимо этого, известны и другие признаки, например, усиленное разрушение инсулина [23], усиленная секреция глюкагона [24], повышенная выработка глюкозы в печени [24], усиленная продукция катехоламинов и инсулинорезистентность [25]. Эти же факторы, как показывают результаты исследований, являются составной частью гипертиреоза [26]. Таким образом, наблюдается ряд физиологических отклонений, общих для гипертиреоза и СД 2 типа. Из вышеперечисленной симптоматологии наиболее важным аспектом, связывающим тиреоидную дисфункцию и СД 2 типа, считается инсулинорезистентность. Это состояние возникает как при гипертиреозе, так и при гипотиреозе [27].
Резистентность к инсулину в мышцах и печени – характерный признак СД 2 типа. Для поддержания нормального уровня глюкозы в крови требуются, прежде всего, ненарушенный гомеостаз глюкозы и интактный секреторный инсулиновый ответ, а также ненарушенная чувствительность тканей к инсулину [28-31].
Утилизация глюкозы опосредуется совокупным эффектом инсулина и гипергликемии, который и модулирует все три основополагающих феномена. Во-первых, это снижение эндогенной (печеночной) продукции глюкозы. Во-вторых, это повышенное усвоение (захват тканями) глюкозы (печеночное или висцеральное). В-третьих, это повышение количества рецепторов глюкозы в периферических тканях (скелетных мышцах). Захват глюкозы мышечной тканью регулируется гликолизом и синтезом гликогена. Печеночная инсулинорезистентность характеризуется избыточной выработкой глюкозы, несмотря на гиперинсулинемию натощак, кроме того, повышенная скорость продукции глюкозы в печени считалась центральным модулятором повышенной концентрации глюкозы плазмы натощак (ГПН) у пациентов с СД 2 типа [24]. Если резистентность к инсулину присутствует в постабсорбционном периоде, отмечается повышение усвоения глюкозы в мышцах, но эффективность усвоения при этом снижается. В свете подобных биохимических процессов, пониженное усвоение глюкозы в мышцах и усиленная продукция глюкозы в печени ведут к ухудшению метаболизма глюкозы.
Термином «гармоничный квартет» называют основополагающий патофизиологический механизм развития инсулинорезистентности [24]. Дестабилизированная утилизация и метаболизм глюкозы в адипоцитах, мышцах и печени, наряду с нарушенной секрецией инсулина бета-клетками поджелудочной железы, представляют собой четыре основных нарушения, которые играют определяющую роль в патогенезе СД 2 типа. При этом следует учитывать, что развитие инсулинорезистентности доказано и при гипертиреозе, и при гипотиреозе. Кроме этого, как показывают последние данные, инсулинорезистентность ведет и к нарушению метаболизма липидов [32]. Таким образом, выходит, что инсулинорезистентность может оказаться связующим звеном между СД 2 типа и тиреоидной дисфункцией.
Инсулинорезистентность и функция бета-клеток обратно коррелируют с тиреотропным гормоном, что может объясняться антагонистическим эффектом тиреоидных гормонов в отношении инсулина наряду с повышением концентрации ТТГ. Повышенный сывороточный уровень ТТГ часто соответствует пониженному содержанию тиреоидных гормонов посредством механизма отрицательной обратной связи. По мере повышения концентрации ТТГ уровень тиреоидных гормонов снижается, а антагонистический эффект в отношении инсулина ослабевает. Эти наблюдения показывают, что дисбаланс инсулина тесно связан с дисфункцией щитовидной железы, и этот феномен обусловлен дисфункцией бета-клеток [33].
Инсулинорезистентность при гипертиреозе и субклиническом гипертиреозе
Гипертиреоз ассоциируется с инсулинорезистентностью, которая, в свою очередь, связана с повышенным метаболизмом глюкозы, усиленной абсорбцией глюкозы в кишечнике, увеличенной продукцией глюкозы в печени, высокими концентрациями свободных жирных кислот, повышенным уровнем инсулина и проинсулина натощак и после приема пищи, а также ускоренным периферическим транспортом глюкозы, сопровождающимся утилизацией глюкозы [27, 34]. Доказано, что пациенты с СД 2 типа и тиреоидной дисфункцией более подвержены кетозу и кетогенезу [35, 36].
|
Клинический случай 2 Пациентка 23 лет, страдает диабетом 1 типа в течение 8 лет, диабет компенсирован (HbA1с <7,5 % на протяжении всего периода болезни, эпизоды ДКА отсутствовали). Выкуривала по 20 сигарет в день, но алкоголь употребляла только изредка. 12 месяцев назад поступила с типичными симптомами тиреотоксикоза и ухудшением гликемического контроля в течение 2 месяцев: ТТГ <0,06 мЕд/л, общий тироксин 196 ммоль/л, антитела к тиреоидной пероксидазе положительные. После лечения КАРБИМАЗОЛОМ у пациентки восстановился гликемический контроль и эутиреоидное состояние на протяжении всего периода приема КАРБИМАЗОЛА (10 мг в сутки). На момент поступления в отделение пациентка в течение недели отмечала слабость, ощущение сердцебиения и ухудшение гликемического контроля, несмотря на строгое соблюдение режима лечения инсулином и диеты. Пациентка была обезвожена, отмечалась гипотензия (90/70 мм рт. ст.), тахикардия (120 уд/мин), симптомы инфекции мочевыводящих и дыхательных путей отрицала. Отмечалась тяжелая глюкозурия, кетонурия, глюкоза капиллярной крови 24 ммоль/л, рН артериальной крови ‒ 7,26, уровень бикарбоната в крови ‒ 12 ммоль/л. Развернутый общий анализ крови, печеночные трансаминазы, почечные показатели, электролиты сыворотки (калий 4,8 ммоль/л), рентгенография грудной клетки и электрокардиограмма ‒ в норме. Кровь и моча были стерильны. Однако ТТГ был понижен на фоне повышения тироксина плазмы (192 нмоль/л). Позднее пациентка призналась, что прекратила принимать КАРБИМАЗОЛ 10 недель назад, будучи в отпуске. Пациентка получала внутривенно жидкости, инсулин и перорально пропранолол и КАРБИМАЗОЛ (30 мг в сутки). Состояние пациентки заметно улучшилось, и через 5 дней она была выписана домой. |
Роль печени
При гипертиреозе эндогенная продукция глюкозы у человека увеличивается, а чувствительность тканей печени к инсулину снижается [40] ввиду гликогенеза и гликогенолиза. Была выдвинута гипотеза о влиянии гипоталамуса на печень через активацию симпатической нервной системы [41], которая наряду с повышенной экспрессией белков-транспортеров ГЛЮТ-2 в печени рано или поздно ведет к повышению плазменных концентраций свободных жирных кислот [42, 43].
|
От редакции КАРБИМАЗОЛ является препаратом первой линии в лечении гипертиреоза практически у всех пациентов, поскольку:
Australian Family Physician Vol. 41, No. 8, Аugust 2012 |
Роль скелетных мышц
При гипертиреоидном состоянии наблюдается заметно усиленная утилизация глюкозы в скелетных мышцах [34]. По имеющимся данным, повышенная утилизация глюкозы опосредована интенсивностью окисления глюкозы, стимулированного инсулином [44-46]. В этих условиях отмечается подавленный гликогенез в силу стимулированной инсулином неокислительной утилизации глюкозы, которая сопровождается перенаправлением внутриклеточной глюкозы в сторону гликолиза и образования лактата [27]. Транспорт лактата от периферических тканей к печени ведет к усиленной продукции глюкозы в рамках цикла Кори. Помимо этого, гипертиреоз ассоциируется со сниженной чувствительностью к инсулину [47]. Повышенную периферическую инсулинорезистентность связывали с усиленной экспрессией биоактивных воспалительных медиаторов, в том числе адипокинов (ИЛ-6 и ФНО-альфа) [16], которые ведут к развитию инсулинорезистентности.
Генетические причины сахарного диабета 2 типа и тиреоидной дисфункции: возможные точки пересечения
Влияние тиреоидных гормонов на печень: роль генов
Выявлены многочисленные гены, которые идентифицируются с глюконеогенезом, метаболизмом гликогена и действием инсулина. К их числу относятся гены глюкозо-6-фосфата, протеинкиназы В (Akt2), β2-адренергического рецептора, ингибирующего G-белка (Gi), фосфоэнолпируваткарбоксилазы (ФЭП-карбоксилазы) [25], пируваткарбоксилазы (ПК), ГЛЮТ-2 [42, 64], малатдегидрогеназы («яблочного фермента») [65] и белка, связывающего углеводчувствительный элемент (ChREBP) [66]. У гипертиреоидных крыс наблюдалась повышенная печеночная экспрессия белка ГЛЮТ-2, по сравнению с гипотиреоидными крысами [64].
По некоторым данным, у мышей с гиперинсулинемией или инсулинорезистентностью повышалась транскрипция различных ферментов, вовлеченных в метаболизм липидов [2, 67].
Влияние тиреоидных гормонов на скелетные мышцы
К числу многочисленных генов, которые влияют на взаимодействие тиреиодных гормонов и скелетных мышц, относятся гены ГЛЮТ-1, ГЛЮТ-4 [64], β2-адренергического рецептора [69], фосфоглицераткиназы (ФГК) [70], РАПП гамма-коактиватора 1-альфа (PGC-1 альфа) [71] и митохондриальных разобщающих белков (РБ) [72]. Из целого ряда выявленных генов подробно изучались гены ГЛЮТ-4 и РБ-3. Как было доказано, в скелетных мышцах ГЛЮТ-4 опосредуется влиянием гормона Т3 и может повышать базальный и инсулинопосредованный транспорт глюкозы [64].
Недавно был открыт ген митохондриального РБ-3. Впервые было показано, что этот ген ассоциируется с метаболизмом глюкозы и снижением окисления жирных кислот [73]. Кроме того, сообщалось, что этот ген играет центральную роль в отрицательно регулируемой активации сигнального пути PKB/Akt и 5' АМФК [2, 73]. Изучалась также роль гормона Т2, который, как было доказано, ассоциируется с сарколеммным ГЛЮТ-4. Фосфофруктокиназа и гликолитические ферменты связаны с Т2-опосредованной активностью ГЛЮТ-4 [74]. Ряд известных генов ассоциируется с периферическим метаболизмом глюкозы [2].
Считается, что в основе генетических нарушений у пациентов с СД, страдающих также от тиреоидной патологии, лежат аутоиммунные причины. Эти сведения поддерживают огромное количество клинических свидетельств связи между СД 1 типа и аутоиммунной дисфункцией щитовидной железы (АДЩЖ) [75, 76]. Множество генов, вовлеченных в метаболизм глюкозы, модулируются активным щитовидным гормоном Т3 путем связывания с рецепторами тиреоидного гормона. Речь идет о рецепторах TR?1, TR?1, TR?2 и TR?3, т.е. о четырех основных изоформах рецепторов, связывающих гормон щитовидной железы [77]. Предполагается, что TR?1 регулирует метаболические эффекты тиреоидного гормона. Рецепторы TR?1 и TR?2 связывают с поддержкой функционирования гипоталамо-гипофизарно-тиреоидной оси и поддержанием эутиреоидного состояния [78].
Исследования показали, что 3,5,3-трииодотиронин является производным от гормона Т4. Он может активироваться путем удаления атома йода из фенольного кольца с помощью деиодиназы иодотиронина 1 (D1) и 2 типа (D2). Деиодиназа иодотиронина 3 типа (D3) инактивирует тиреоидный гормон путем удаления атома йода из тирозильного кольца. Деиодиназы экспрессируются в различных тканях, и уровень их экспрессии колоссально варьируется в процессе развития, а регуляция осуществляется состоянием гормонов щитовидной железы. D1 экспрессируется в печени, а D2 – в жировой ткани и скелетных мышцах. Они участвуют в регуляции биодоступности Т3 и, следовательно, – ответа на инсулин. Повышенная концентрация Т3 ассоциируется с новейшей миссенс-мутацией (Thr92Ala). Этот феномен тесно связан с инсулинорезистентностью. Это также связано с резкими колебаниями метаболизма глюкозы, что сопровождается повышающей up-регуляцией инсулинопосредованной утилизации глюкозы в скелетных мышцах и жировой ткани.
Это явление, опосредованное положительной регуляцией чувствительной к инсулину транскрипции белка ГЛЮТ-4 [78, 79], указывает на существование глубинных геномных эффектов гормона Т3 на метаболизм глюкозы в печени. Рецептор тиреоидного гормона (TR), экспрессирующийся в гепатоцитах, и стимуляция Т3-чувствительных нейронов в ходе модулированной гипоталамусом продукции глюкозы в печени посредством симпатических проекций в печени опосредуются циркулирующими глюкорегуляторными гормонами [79]. Недавно полученные данные, пролившие свет на полиморфизм гена деиодиназы 2 типа (DIO2), Thr92Ala, дают основания говорить о гомозиготности для этого полиморфизма, который, в свою очередь, отвечает за повышенный риск развития СД 2 типа [80].
Отдельныйы метаанализ показал, что внутриклеточный трийодтиронин (Т3) отвечает за отклонения в чувствительности к инсулину [4]. Кроме того, сообщалось, что полиморфизм Thr92Ala приводит к снижению активности деиодиназы 2 типа, что, в свою очередь, ассоциировалось с резистентностью к инсулину. Ген D2 характеризуется своеобразной транскрипционной и посттрансляционной регуляцией. Это потенциальный модулятор действия инсулина в скелетных мышцах и жировой ткани, активность которого проявляется посредством регуляции транскрипции гена ГЛЮТ-4 [81].
Тиреотоксикоз вызывает усиленное перекисное окисление липидов. Клиренс липопротеинов низкой плотности (ЛПНП) ведет к снижению уровня холестерина и триглицеридов. ТГ провоцирует повышающую регуляцию эффектов катехоламинов, что приводит к липолизу в адипоцитах и повышению уровня циркулирующих в крови ЖК. Повышенное количество ЖК компенсирует ТГ-опосредованное усиленное окисление длинноцепочечных ЖК в печени. Повышенный уровень циркулирующих ЖК и наличие гликонеогенных субстратов из периферических резервов вызывают ответный повышенный глюконеогенез у животных, леченных T3. По имеющимся данным, Т3 повышает содержание глюкозы в плазме крови натощак и концентрацию свободных ЖК. Активацией периферических субстратов объясняется стремительный скачок гипергликемии при тиреотоксикозе [84]. Парадоксально, но гипергликемический эффект тиреотоксикоза может сохраняться на фоне повышенного кровоснабжения мышц, учитывая усиленное поступление субстрата из тканей [16]. Этот феномен отображен на рисунке 2.
|
Печень |
|
Усиленный глюконеогенез |
|
Липогенез |
|
Сниженный синтез гликогена |
|
Повышенная продукция глюкозы |
|
Периферические ткани |
|
Ускоренный транспорт глюкозы |
|
Повышенный липолиз |
|
Повышенный гликолиз |
|
Усиление функции митохондрий |
Рис. 2. Влияние гормонов щитовидной железы на печень и периферические ткани
Клинические руководства, регулирующие место диагностики щитовидной дисфункции у пациентов с сахарным диабетом 2 типа
Для понимания роли, важности и необходимости выявления дисфункции щитовидной железы у пациентов с СД 2 типа были проведены многочисленные исследования. Бесспорной и очевидной оказалась необходимость назначения обследования на предмет выявления дисфункции щитовидной железы у пациентов с СД 2 типа, причем с ежегодным его повторением [13]. Рекомендации по скринингу щитовидной железы у пациентов с СД в Великобритании и США представлены в таблице 1.
Таблица 1. Практические руководства по скринингу функции щитовидной железы у пациентов с сахарным диабетом
|
N |
Руководство |
Сахарный диабет 2 типа |
Комментарии |
|
1 |
Руководство Американской тиреоидной ассоциации; выявление дисфункции щитовидной железы [61] |
Пациентам с сахарным диабетом могут требоваться более частые обследования |
Рекомендуется проверять уровень ТТГ у всех взрослых, начиная с 35 лет, каждые 5 лет; людям из группы высокого риска анализы можно назначать чаще. Диабет указывается как повышенный фактор риска, разграничение между СД 1 и 2 типа не проводится. |
|
2 |
Американская ассоциация клинических эндокринологов; клинические протоколы при заболеваниях щитовидной железы от 2002 г. [62] |
Пальпация щитовидной железы и анализ на ТТГ при постановке диагноза и далее с регулярными интервалами, особенно при зобе и других аутоиммунных заболеваниях |
Специфических рекомендаций для СД 2 типа нет.
|
|
3 |
Руководство Британской тиреоидной ассоциации и Ассоциации клинической биохимии от 2006 г. [63] |
Исследование функции щитовидной железы на исходном уровне, однако ежегодные анализы не рекомендуются. |
У пациенток с СД во время беременности и после родов рекомендуется проводить анализы на ТТГ и антитела. |
Примечания: СД ‒ сахарный диабет; ТТГ ‒ тиреотропный гормон.
Руководство Американской тиреоидной ассоциации для пациентов с СД 2 типа требуют частого проведения анализов на функцию щитовидной железы. Согласно этим рекомендациям, анализы следует проводить всем взрослым каждые 5 лет, начиная с 35 лет. Пациентам из группы высокого риска анализы могут назначаться чаще. Клинические протоколы при заболеваниях щитовидной железы от Американской ассоциации клинических эндокринологов (2002 г.) рекомендуют выполнять пальпацию щитовидной железы и анализ на ТТГ, особенно если зоб и другие аутоиммунные заболевания возникают на фоне СД 2 типа. Регулярный скрининг патологии щитовидной железы у всех пациентов с СД позволит начать раннее лечение субклинической дисфункции щитовидной железы. Чувствительный анализ на ТТГ в сыворотке крови считается скрининговым тестом выбора. Кроме того, пациентам с СД 2 типа предлагалось выполнять анализ на ТТГ на момент постановки диагноза и далее не реже одного раза каждые 5 лет.
|
Обсуждение двух клинических случаев Диабетический кетоацидоз (ДКА) характеризуется тяжелым нарушением метаболизма углеводов, белков и липидов, главным образом в результате отсутствия или неэффективности инсулина, на фоне сопутствующего повышения антагонистичных гормонов (глюкагона, катехоламинов и кортизола). Провоцирующий фактор не всегда удается установить. В отсутствие любых других факторов, которые так и не удалось обнаружить после тщательного исследования, мы считаем, что причиной ухудшения гликемического статуса и развития кетоацидоза у обоих наших пациентов был тиреотоксикоз. В работе Nijs было показано, что повышенный клиренс инсулина у пациентов с инсулинозависимым сахарным диабетом и тиреотоксикозом возвращается в норму при улучшении тиреотоксического статуса. Важно!
|
Выводы
В лечении заболеваний внутренних органов неоднократно было показано, что связь между дисфункцией щитовидной железы и СД очевидна. Хорошо известно, что при тиреотоксикозе часто развиваются изменения в метаболизме. У людей, не страдающих диабетом, скачки глюкозы в плазме крови и инсулиновый ответ обычно находятся в пределах нормы, однако примерно у каждого третьего человека наблюдается нарушение толерантности к глюкозе и недостаточный инсулиновый ответ на глюкозную нагрузку. При тиреотоксикозе же возрастает степень всасывания глюкозы и ее продукция из гликогена, а также повышаются уровни лактата, глицерина и аминокислот. Срыв компенсации диабета при тиреотоксикозе может быть связан с увеличением базальной продукции глюкозы в печени и снижением потенциала ее подавляемости под действием инсулина. Другие механизмы включают повышенную резистентность периферических тканей к инсулину и повышенный клиренс инсулина. Инсулинорезистентность при тиреотоксикозе может быть следствием скорее повышенной продукции глюкозы в печени, чем пострецепторного дефекта. Таким образом, при тиреотоксикозе могут наблюдаться нарушения в секреции инсулина, продукции глюкозы в печени, ее подавляемости под действием инсулина, чувствительности к инсулину периферических тканей и распаде инсулина. Очень важно диагностировать дисфункцию щитовидной железы у пациентов с СД 2 типа, и это положение следует внедрять в клиническую практику, что, наверняка, немедленно повлияет на более качественное понимание связи между нарушением функции щитовидной железы с СД 2 типа.
Список литературы:
- F. A. Coller and C. B.Huggins, “Effect of hyperthyroidism upon diabetes mellitus: striking improvement in diabetes mellitus from thyroidectomy,” Annals of Surgery, vol. 86, no. 6, pp. 877–884, 1927.
- G. Brenta, S. Danzi, and I. Klein, “Potential therapeutic applications of thyroid hormone analogs,” Nature Clinical Practice Endocrinology and Metabolism, vol. 3, no. 9, pp. 632–640, 2007.
- F. Goglia, M. Moreno, and A. Lanni, “Action of thyroid hormones at the cellular level: the mitochondrial target,” FEBS Letters, vol. 452, no. 3, pp. 115–120, 1999. Journal of Diabetes Research 7
- R. Kadiyala,R.Peter, andO. E. Okosieme, “Thyroiddysfunction in patients with diabetes: clinical implications and screening strategies,” International Journal of Clinical Practice, vol. 64, no. 8, pp. 1130–1139, 2010.
- O. Kordonouri, A. M. Maguire, M. Knip et al., “Other complications and associated conditions with diabetes in children and adolescents,” Pediatric Diabetes, vol. 10, no. 12, pp. 204–210, 2009.
- R. W. Holl, B. B¨ohm, U. Loos, M. Grabert, E. Heinze, and J. Homoki, “Thyroid autoimmunity in children and adolescents with type 1 diabetes mellitus. Effect of age, gender and HLA type,” Hormone Research, vol. 52, no. 3, pp. 113–118, 1999.
- J. M. Barker, J.Yu, L. Yu et al., “Autoantibody ”subspecificity” in type 1 diabetes: risk for organ-specific autoimmunity clusters in distinct groups,” Diabetes Care, vol. 28,no. 4, pp. 850–855, 2005.
- J. E.Donckier, “Endocrine diseases and diabetes,” in Text Book of DiabetesMellitus, J. C. Pickup andG.Williams, Eds., vol. 27, pp. 21–27, Blackwell Publishing Company, Chichester, UK, 2003.
- J. D. Baxter, W. H. Dillmann, B. L. West et al., “Selective modulation of thyroid hormone receptor action,” Journal of Steroid Biochemistry and Molecular Biology, vol. 76, no. 1–5, pp. 31–42, 2001.
- J.D. Baxter and P.Webb, “Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes,” Nature Reviews Drug Discovery, vol. 8, no. 4, pp. 308–320, 2009.
- W. M. G. Tunbridge,D. C. Evered, and R.Hall, “Thespectrumof thyroid disease in a community: theWhickhamsurvey,” Clinical Endocrinology, vol. 7, no. 6, pp. 481–493, 1977.
- J. G. Hollowell, N. W. Staehling, W. D. Flanders et al., “Serum TSH, T4, and thyroid antibodies in theUnited States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III),” Journal of Clinical Endocrinology and Metabolism, vol. 87, no. 2, pp. 489–499, 2002.
- P. Perros, R. J. McCrimmon, G. Shaw, and B. M. Frier, “Frequency of thyroid dysfunction in diabetic patients: value of annual screening,” DiabeticMedicine, vol. 12, no. 7, pp. 622–627, 1995.
- D. H. Akbar, M. M. Ahmed, and J. Al-Mughales, “Thyroid dysfunction and thyroid autoimmunity in Saudi type 2 diabetics,” Acta Diabetologica, vol. 43, no. 1, pp. 14–18, 2006.
- S. Stanick´a, K. Vondra, T. Pelik´anov´a, P. Vlˇcek, M. Hill, and V. Zamrazil, “Insulin sensitivity and counter-regulatory hormones in hypothyroidism and during thyroid hormone replacement therapy,” Clinical Chemistry and Laboratory Medicine, vol. 43, no. 7, pp. 715–720, 2005.
- P. Mitrou, S. A. Raptis, and G. Dimitriadis, “Insulin action in hyperthyroidism: a focus on muscle and adipose tissue,” Endocrine Reviews, vol. 31, no. 5, pp. 663–679, 2010.
- V. Lambadiari, P.Mitrou, E. Maratou et al., “Thyroid hormones are positively associated with insulin resistance early in the development of type 2 diabetes,” Endocrine, vol. 39, no. 1, pp. 28–32, 2011.
- M. Potenza, M. A. Via, and R. T. Yanagisawa, “Excess thyroid hormone and carbohydrate metabolism,” Endocrine Practice, vol. 15, no. 3, pp. 254–262, 2009.
- M. S. Eledrisi, M. S. Alshanti, M. F. Shah, B. Brolosy, and N. Jaha, “Overview of the diagnosis and management of diabetic ketoacidosis,”American Journal of theMedical Sciences, vol. 331, no. 5, pp. 243–251, 2006.
- T. L. Althausen and M. Stockholm, “The influence of the thyroid gland on absorption in the digestive tract,” The American Journal of Physiology, vol. 123, no. 3, pp. 577–588, 1938.
- L. H. Duntas, J. Orgiazzi, and G. Brabant, “The interface between thyroid and diabetes mellitus,” Clinical Endocrinology, vol. 75, no. 1, pp. 1–9, 2011.
- A. Clark, L. C. Jones, E. de Koning, B. C. Hansen, and D. R. Matthews, “Decreased insulin secretion in type 2 diabetes: a problem of cellular mass or function?” Diabetes, vol. 50, no. 1, pp. S169–S171, 2001.
- J. P. Randin, B. Scazziga, E. Jequier, and J. P. Felber, “Study of glucose and lipid metabolism by continuous indirect calorimetry in Graves’ disease: effect of an oral glucose load,” Journal of Clinical Endocrinology and Metabolism, vol. 61, no. 6, pp. 1165–1171, 1985.
- R. A. DeFronzo, “Pathogenesis of type 2 diabetes mellitus,” Medical Clinics of North America, vol. 88, no. 4, pp. 787–835, 2004.
- D. M. Muoio and C. B. Newgard, “Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and ?-cell failure in type 2 diabetes,” Nature Reviews Molecular Cell Biology, vol. 9, no. 3, pp. 193–205, 2008.
- G. Dimitriadis, P. Mitrou, V. Lambadiari et al., “Insulin-stimulated rates of glucose uptake in muscle in hyperthyroidism: the importance of blood flow,” Journal of Clinical Endocrinology and Metabolism, vol. 93, no. 6, pp. 2413–2415, 2008.
- G. Brenta, F. S. Celi, M. Pisarev, M. Schnitman, I. Sinay, and P. Arias, “Acute thyroid hormone withdrawal in athyreotic patients results in a state of insulin resistance.,” Thyroid, vol. 19, no. 6, pp. 665–669, 2009.
- R. A. DeFronzo, R. Gunnarsson, and O. Bjorkman, “Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus,” Journal of Clinical Investigation, vol. 76, no. 1, pp. 149–155, 1985.
- R. A.DeFronzo, “Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes genes,” Diabetes Reviews, vol. 5, no. 3, pp. 177–269, 1997.
- A.Mari, J.Wahren, R. A.DeFronzo, and E. Ferrannini, “Glucose absorption and production following oral glucose: comparison of compartmental and arteriovenous-difference methods,” Metabolism: Clinical and Experimental, vol. 43, no. 11, pp. 1419–1425, 1994.
- L. Mandarino, R. Bonadonna, O. McGuinness, and D. Wasserman, “Regulation of muscle glucose uptake in vivo,” in Handbook of Physiology, The Endocrine System, The Endocrine Pancreas and Regulation of Metabolism, L. S. Jefferson and A. D. Cherrington, Eds., vol. 2, pp. 803–848, Oxford University Press, Oxford, UK, 2001.
- K. B. Kapadia, P. A. Bhatt, and J. S. Shah, “Association between altered thyroid state and insulin resistance,” Journal of Pharmacology and Pharmacotherapeutics, vol. 3, pp. 156–160, 2012.
- G. Chen, J. Wu, Y. Lin et al., “Associations between cardiovascular risk, insulin resistance, ?-cell function and thyroid dysfunction: a cross-sectional study in She ethnic minority group of Fujian Province in China,” European Journal of Endocrinology, vol. 163, no. 5, pp. 775–782, 2010.
- G. D. Dimitriadis and S. A. Raptis, “Thyroid hormone excess and glucose intolerance,” Experimental and Clinical Endocrinology and Diabetes, vol. 109, no. 2, pp. S225–S239, 2001.
- M. Beylot, J. P. Riou, F. Bienvenu, and R. Mornex, “Increased ketonaemia in hyperthyroidism. Evidence for a ?-adrenergic mechanism,” Diabetologia, vol. 19, no. 6, pp. 505–510, 1980.
- M. Beylot, “Regulation of in vivo ketogenesis: role of free fatty acids and control by epinephrine, thyroid hormones, insulin 8 Journal of Diabetes Research and glucagon,” Diabetes andMetabolism, vol. 22, no. 5, pp. 299–304, 1996.
- J. Rezzonico, H. Niepomniszcze, M. Rezzonico, E. Pusiol, M. Alberto, and G. Brenta, “The association of insulin resistance with subclinical thyrotoxicosis,” Thyroid, vol. 21, no. 9, pp. 945– 949, 2011.
- M. Peppa, G. Betsi, and G. Dimitriadis, “Lipid abnormalities and cardiometabolic risk in patients with overt and subclinical thyroid disease,” Journal of Lipids, vol. 2011, Article ID 575840, 9 pages, 2011.
- C. C. Wang and J. E. B. Reusch, “Diabetes and cardiovascular disease: changing the focus fromglycemic control to improving long-term survival,” The American Journal of Cardiology, vol. 110, no. 9, pp. 58B–68B, 2012.
- P. Cavallo-Perin, A. Bruno, L. Boine, M. Cassader, G. Lenti, and G. Pagano, “Insulin resistance in Graves’ disease: a quantitative in-vivo evaluation,” European Journal of Clinical Investigation, vol. 18, no. 6, pp. 607–613, 1988.
- L. P. Klieverik, C. P. Coomans, E. Endert et al., “Thyroid hormone effects on whole-body energy homeostasis and tissuespecific fatty acid uptake in vivo,” Endocrinology, vol. 150, no. 12, pp. 5639–5648, 2009.
- S. P. Weinstein, E. O’Boyle, M. Fisher, and R. S. Haber, “Regulation of GLUT2 glucose transporter expression in liver by thyroid hormone: evidence for hormonal regulation of the hepatic glucose transport system,” Endocrinology, vol. 135, no. 2, pp. 649–654, 1994.
- T.Mokuno,K.Uchimura,R.Hayashi et al., “Glucose transporter 2 concentrations in hyper- and hypothyroid rat livers,” Journal of Endocrinology, vol. 160, no. 2, pp. 285–289, 1999.
- G. Dimitriadis, B. Baker, H. Marsh et al., “Effect of thyroid hormone excess on action, secretion, andmetabolism of insulin in humans.,” The American Journal of Physiology, vol. 248, no. 5, pp. E593–601, 1985.
- A. J. McCulloch, D. G. Johnston, and P. H. Baylis, “Evidence that thyroid hormones regulate gluconeogenesis from glycerol in man,” Clinical Endocrinology, vol. 19, no. 1, pp. 67–76, 1983.
- M. C. Foss, G.M. G. F. Paccola,M. J. A. Saad,W. P. Pimenta, C. E. Piccinato, and N. Iazigi, “Peripheral glucose metabolism in human hyperthyroidism,” Journal of Clinical Endocrinology and Metabolism, vol. 70, no. 4, pp. 1167–1172, 1990.
- D.C. Shen,M. B. Davidson, S.W.Kuo, andW. H. Sheu, “Peripheral and hepatic insulin antagonism in hyperthyroidism,” Journal of Clinical Endocrinology and Metabolism, vol. 66, no. 3, pp. 565–569, 1988.
- M. Haluzik, J. Nedvidkova, V. Bartak et al., “Effects of hypo and hyperthyroidism on noradrenergic activity and glycerol concentrations in human subcutaneous abdominal adipose tissue assessed with microdialysis,” Journal of Clinical
Опубликовано в научно-медицинском журнале «ДОМС»
